本文发表于《大众科学》的前博客网络,仅反映作者的观点,不一定反映《大众科学》的观点
微生物无处不在,更不用说在我们体内了,但是发现和描述它们并非易事。但事情正在变得容易。正如我去年写到的,DNA测序技术的进步意味着我们不再局限于分离单个微生物,在实验室中培养它们并逐个研究。相反,我们可以从特定环境中分离DNA,然后对其中存在的所有物质进行测序。但是,新期刊《自然·微生物学》上发表的一篇论文表明,分析新微生物种群的最常用方法系统地遗漏了其中很大一部分。
除非您是微生物学家,否则您可能需要了解标题中的两个术语——“宏基因组学”和“基于扩增子的检测”的入门知识。我在去年的系列文章中对它们进行了更完整的介绍,但我将简要回顾一下。对于这种测序方法,一个有用的比喻是走进图书馆并尝试编目其内容。扩增子测序就像扫描书脊并仅记录其标题,而宏基因组测序就像浏览并记录所有书籍内的文本。当然,您可以想象哪种方法更容易。
关于支持科学新闻报道
如果您喜欢这篇文章,请考虑通过以下方式支持我们屡获殊荣的新闻报道 订阅。通过购买订阅,您正在帮助确保有关塑造我们当今世界的发现和想法的具有影响力的故事的未来。
这个比喻并不完美,因为基因组没有容易访问的“标题”。相反,科学家们知道标题通常采用的形式,并且可以使用称为聚合酶链反应或PCR的技术来扩增这些序列。这篇论文表明,科学家们一直在使用的所谓“通用”引物并不像我们想象的那么通用。这就像我们只一直在寻找以字母开头的标题,并且在许多情况下完全错过了奥威尔的《1984》。
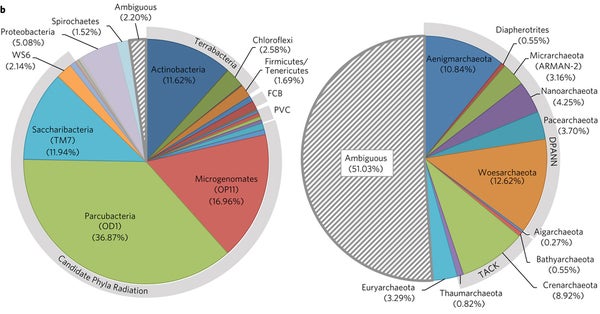
Emiley A. Eloe-Fadrosh 及其同事分析了超过 6000 个宏基因组数据集,并检查了常用微生物引物可以检测到多少 SSU 基因(用作基因组标题的基因)。他们发现,10% 或更多会被完全遗漏。毫不奇怪,大多数会被遗漏的细菌是新型或至少研究非常不足的物种的成员。

图 1a - 给定引物会遗漏的宏基因组样本中微生物的百分比。
不幸的是,解决这个问题的方法并不明显。作者指出了单细胞和深度宏基因组测序,但是这些技术仍然相对昂贵,尤其是在分析返回的大量数据所需的专业知识和计算基础设施方面。在功能性地描述这些大量的新细菌方面也存在巨大差距——仅仅因为我们知道它们在那里,并不意味着我们知道它们是做什么的。尽管如此,知道它们在那里是必要的第一步。