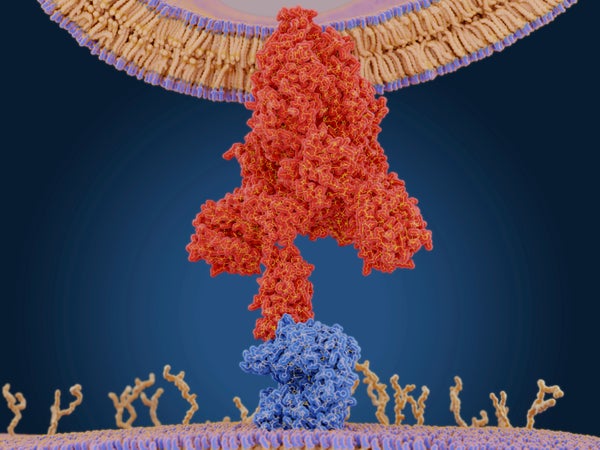
当病毒入侵你的细胞时,它会改变你的身体。但在此过程中,病原体也会改变其形状。一种新的数学模型预测了病毒上允许这种形状转变发生的点,揭示了一种寻找潜在药物和疫苗靶点的新方法。这种独特的基于数学的方法已经确定了引起 COVID-19 的冠状病毒中的潜在靶点。
该策略在 4 月份发表于《计算生物学杂志》,预测了病毒上的蛋白质位点,这些位点储存着能量——重要的药物可以抑制的部位。该研究的作者、法国高等科学研究所的数学家罗伯特·彭纳说,这项工作罕见地从纯数学出发。“在生物学中,纯数学非常少见,”他补充说。威斯康星大学麦迪逊分校研究病毒的约翰·尹说,这篇论文的预测在实验验证之前还有很长的路要走,他没有参与这项研究。但他同意彭纳的方法具有潜力。“他是从数学家的角度来看待这个问题——但却是一位非常了解科学的数学家,”尹说。“所以这非常罕见。”
彭纳的方法利用了一个事实,即某些病毒蛋白在病毒突破细胞时会急剧改变其形状,而这种转变取决于不稳定的特征。(根据定义,稳定的蛋白质位点会抵抗变化。)通过识别“高自由能位点”——病毒蛋白上储存大量能量的区域——彭纳意识到他可以找到可能介导这种形状变化的“弹簧”点。他称这种高能点为奇异位点。找到它们需要一些复杂的数学。
支持科学新闻
如果您喜欢这篇文章,请考虑通过 订阅来支持我们屡获殊荣的新闻报道。通过购买订阅,您将帮助确保关于塑造我们当今世界的发现和想法的有影响力的故事的未来。
彭纳专注于细胞融合和进入过程中变化最大的蛋白质的骨架。他检查了蛋白质折叠时在骨架部分之间形成的氢键。蛋白质由一系列独立的单元(或残基)组成,其中两个这样的单元形成氢键。键合的单元相对于彼此旋转,这些扭曲意味着所涉及的残基中存在不同数量的自由能。
为了分离出奇异的旋转,彭纳在大量蛋白质形状上应用了一些数学方法。他和他的同事之前从数据库中收集了具有代表性的蛋白质样本,并查看了该组中大约 117 万个骨架氢键。然后,他需要确定不同旋转出现的频率。
为了找到这些信息,彭纳转向了几何学。在 19 世纪,德国数学家卡尔·弗里德里希·高斯证明了可以通过指定旋转围绕其旋转的轴以及旋转的量来描述三维空间的每个独特旋转(想象一下车轮围绕车轴旋转,从零到 360 度,或零到两 pi 弧度)。您可以使用一个向量来表示每次旋转,向量是一种既有大小又有方向的测量值,通常被描绘成指向特定方向的具有一定长度的箭头。这个箭头的方向描述了旋转的轴,向量的长度给出了旋转的量(想象一下轴随着进一步旋转而变长)。从中心点收集所有指向各个方向的向量箭头,您就拥有了旋转可以围绕其旋转的所有可能轴。沿每个轴的点(不同向量的箭头点)标识了所有独特的旋转:从零到两 pi 弧度,围绕每个轴可能的旋转量。
总而言之,这些箭头构成了一个 3D 球(想象一个带刺的 Koosh 球或一个卷起来的刺猬)。这个结构正是彭纳想要的,因为它允许他对其中出现的点进行一些数学运算。彭纳将数据库中发现的旋转映射到球体上。然后,他通过查看其在结构中周围区域的密度来计算每个旋转的频率:球体中密度较低部分的旋转更为罕见。
科学家们知道蛋白质特征的频率与其自由能的函数相关,稀有的特征具有更高的能量。因此,利用已建立的方程和球体上的密度,彭纳计算了不同旋转的自由能,从而揭示了奇异位点。彭纳说,这种方法有效的一个迹象是它预测了已经已知的功能位点。但这种方法发现的以前未知的位点可能被证明是很有希望的药物攻击靶点。
如果实验验证了彭纳预测的位点(如果这是一个很大的问题),那么这种方法很有前景,法国国家科学研究中心的生物学研究员、数学家的顾问阿恩特·贝内克说。“如果是这样,那么自由能自然会成为我们目前没有针对的目标,”他说。“关于药物或抗体可以或应该做什么的整个想法可能会发生改变。”
在周三发表在同一杂志上的后续研究中,彭纳确定了 COVID-19 背后的冠状病毒上的三个奇异的“感兴趣的位点”。但现在它们必须经受住实验室的严格考验。贝内克说,实验人员需要证明敲除这些位点确实会释放自由能。即使那样,它们也可能仍然无法被药物接触到,他补充说。任何针对这些位点的治疗方法都必须经受住动物模型和人体中疗效和安全性的常规测试。“文献中充斥着失败,”彭纳说。
尽管如此,如果该方法有效,它可能会有更广泛的目标应用,从允许细胞与环境交流的信号蛋白到朊病毒(疯牛病等疾病背后的错误折叠的蛋白质)。“这可能远远超出病毒的范围,”贝内克说。